Химико-биологический факультет
Кафедра химии
ДИПЛОМНАЯ РАБОТА
Изучение ионоселективных и индикаторных свойств халькогенидов свинца в водных и неводных средах
Содержание
1 Теоретические основы исследования ионоселективных и индикаторных свойств халькогенидов свинца
1. 1 Общие сведения о потенциометрии
1. 1. 1 Прямая потенциометрия
1. 1. 2 Потенциометрическое титрование
1. 2 Электроды в потенциометрическом анализе
1. 2. 1 Ионоселективные электроды с твёрдыми мембранами
1. 3 Ионоселективные и индикаторные свойства халькогенидов свинца
2 Экспериментальные методы исследования ионоселективных и индикаторных свойств халькогенидов свинца
2. 1 Физико-химические свойства халькогенидов свинца
2. 1. 1 Сульфид свинца
2. 1. 2 Селенид свинца
2. 1. 3 Теллурид свинца
2. 2 Синтез и идентификация халькогенидов свинца
2. 3 Растворы, установка и методика измерений
2. 4 Изучение ионоселективных свойств
2. 5 Изучение индикаторных свойств
2. 5. 1 Кислотно-основное титрование
Список использованных источников
1 Теоретические основы исследования ионоселективных и индикаторных свойств халькогенидов свинца
1. 1 Общие сведения о потенциометрии
Потенциометрия объединяет методы определения различных физико -химических величин и концентраций веществ, основанные на измерении электродвижущих сил (э. д. с. ) обратимых электрохимических цепей, когда рабочий электрод имеет потенциал, близкий к равновесному значению.
Основы потенциометрии заложены В. Нернстом, который в 1889 г. получил известное уравнение (1) для равновесных электродных потенциалов:
![]()
где E - электродный потенциал; E - стандартный электродный потенциал; z - число электронов, участвующих в процессе; R - универсальная газовая постоянная (R = 8, 31 Дж/(молъхК)); T - абсолютная температура; F - постоянная Фарадея (F = 96485, 35 Клхмолъ-1); aox и aRed - активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции; 2, 3 - коэффициент пересчёта натурального логарифма в десятичный.
Вскоре потенциометрия начала применяться в аналитической химии и в 1893 г. Р. Беренд провёл первое потенциометрическое титрование. В настоящее время потенциометрия широко применяется в аналитической и физической химии [1].
Равновесный потенциал - это разность потенциалов или э. д. с., измеряемая в системе, находящийся в равновесии по отношению к электронам, то есть в такой системе, в которой полностью или практически полностью отсутствует направленное движение электронов в проводниках, потенциалы которых измеряют. Другими словами, равновесные потенциалы измеряют по такой методике, которая не требует ни потребления электрического тока, ни его введения в электрохимическую ячейку. Однако, это лишь означает, что суммарный ток между системой и вешней измерительной цепью равен или близок к нулю, а в самой системе могут иметь место электрические токи [2].
В настоящее время потенциометрия широко используется для определения различных физико-химических величин, в аналитической химии - для определения концентрации веществ в растворах, в автоматических непрерывных методах контроля различных технологических процессов и развивается в нескольких направлениях.
Редоксометрия объединяет ряд методов, основанных на измерении окислительно-восстановительных потенциалов в растворах. Основоположником редоксометрии принято считать В. Кларка, который опубликовал первое исследование в этом направлении в 1923 г.
Редоксометрия в настоящее время широко применяется для изучения протолитических процессов и комплексообразования (в химии), для контроля технологических процессов в фармацевтической, текстильной, гидрометаллургической и ряде других oбластей науки и производства.
Ионометрия - область прямой потенциометрии, которая объединяет методы прямого определения концентрации или активности ионов в различных фазах с использованием ионоселективных электродов. К области ионо-метрии относятся рН-метрия, катионометрия и анионометрия.
Определение термодинамических величин широко используется в физической химии. С помощью потенциометрии определяются изменение величины свободной энергии, энтропии, энтальпии в химических процессах, а также активности (концентрации) в водных и неводных растворах и расплавах.
Потенциометрическое титрование - использование потенциометрических методов для наблюдения за ходом реакции титрования, особенно для нахождения точки эквивалентности.
По сравнению с индикаторным титрованием этот метод позволяет достичь большей точности, а также позволяет титровать вещества, для которых отсутствуют цветные индикаторы или их применение невозможно. Часто за один приём можно определить несколько веществ, присутствующих в растворе.
При потенциометрическом титровании используются реакции нейтрализации, осаждения, комплексообразования, окисления-восстановления [1].
1. 1. 1 Прямая потенциометрия
Потенциометрический анализ основан на измерении потенциала индикаторного электрода, величина которого зависит от концентрации определяемого вещества в растворе.
Измерение потенциала отдельного электрода практически неосуществимо, поэтому в методе прямой потенциометрии в электрохимическую ячейку помещают два электрода и потенциал индикаторного электрода измеряют относительно электрода сравнения, потенциал которого в условиях проведения анализа остается постоянным и не зависит от концентрации определяемого вещества [3].
Во всех методах прямой потенциометрии используется зависимость потенциала индикаторного (как правило, селективного) электрода от активности или концентрации определяемого вещества. Электродвижущая сила гальванической цепи равна:
Е = Еср - Е инд
где Е - э. д. с. элемента, Еср - потенциал электрода сравнения, Еинд - потенциал индикаторного электрода [4].
Существует ряд условий, обязательных при проведении прямых потенциометрических измерений:
- постоянство температуры;
- оптимальное значение pH раствора, что существенно с точки зрения оптимизации параметров электродной функции и сохранения химической формы определяемого вещества;
- оптимальный состав анализируемого раствора с учётом селективности электрода (маскирование мешающих компонентов), а также максимально возможной воспроизводимости, правильности и чувствительности измерений [5].
Уравнение Нернста (1) даёт простое соотношение между потенциалом электрода и активностью определяемого иона в растворе. Поэтому на основании измеренной э. д. с. гальванического элемента, зная потенциал электрода сравнения, можно вычислить потенциал индикаторного электрода, а затем рассчитать активность и концентрацию определяемого иона. Однако здесь возникают некоторые трудности.
Во-первых, реально измеряемая э. д. с. гальванического элемента включает в себя кроме потенциалов электродов и диффузионный потенциал (потенциал жидкостного соединения), который возникает между анализируемым раствором и раствором электрода сравнения. Этот потенциал может достигать десятков милливольт, но точно измерить или оценить его невозможно. Его можно свести к минимуму, используя для соединения между растворами солевой мостик, состоящий из концентрированного раствора электролита, ионы которого имеют одинаковую подвижность, например, насыщенного раствора хлорида калия. При этом потенциал жидкостного соединения составляет обычно несколько милливольт или меньше., то есть имеет несущественную для большинства электроаналитических методов величину стандартного потенциала (или погрешность в измеряемой э. д. с. ) даже на 1 мВ даёт относительную ошибку 4 %.
Во-вторых, для вычисления активности определяемого иона по уравнению Нернста надо знать величину стандартного потенциала индикаторного электрода, однако для многих электродов, в первую очередь это касается мембранных (которые в основном и используются как индикаторные в ионо-метрии), это неизвестный потенциал ассиметрии, величина которого изменяется во времени.
В-третьих, из уравнения Нернста можно определить активность, а для вычисления концентрации определяемого вещества требуется знание коэффициентов активности, которые, как правило, недоступны, поскольку обычно состав, а значит и ионная сила раствора, неизвестны. Поэтому при использовании прямой потенциометрии для аналитических целей применяют, обычно, эмпирическую калибровку измерительного электрода.
Готовят серию стандартных растворов. Измеряют э. д. с. гальванического элемента с каждым из этих растворов и строят калибровочный график зависимости э. д. с. от логарифма активности или концентрации иона. Измерив э. д. с. элемента с анализируемым раствором по графику определяют активность или концентрацию искомого иона. В некоторых случаях, например при определении рН растворов, вместо построения графика настраивают (калибруют) прибор [6].
Для успешного применения метода важно, чтобы ионный состав стандартного раствора был приблизительно таким же, как и состав исследуемого раствора, что трудновыполнимо при анализе сложных образцов. Если концентрация электролита в анализируемом растворе невысока, то полезно разбавить и пробу, и стандартные растворы, используемые для построения градуировочного графика, избытком инертного электролита. В этих условиях добавочное влияние электролита пробы становится незначительным и градуировочный график дает результаты в единицах концентрации. Верхний концентрационный предел нельзя считать серьезным ограничением, так как это ограничение легко устранить путем разбавления исходного раствора до требуемой ионной силы. Чтобы уменьшить различие в ионной силе растворов, можно добавить раствор, регулирующий ионную силу.
Величина pH анализируемого раствора должна быть оптимальной. Доступность и широкая применимость ионоселективных электродов вырабатывают у химика уверенность в правильности любого измерения. Однако для каждой электродной системы имеются ограничения:
- занижение результатов вследствие влияния ОН-ионов;
- занижение и завышение результатов из-за влияния ионов водорода.
Так, в растворах Сu2+ с высокой концентрацией ОН- - ионов возможно образование Сu(ОН)2. Чтобы результаты анализа не оказались заниженными, pH растворов снижают до 8, 0, добавляя HCl. Для определения концентрации ионов натрия используют натрий-селективный электрод, который чувствителен также к ионам водорода. Поэтому результаты определения натрия при низких значениях pH получаются завышенными. Повышая pH раствора до 8, 5 можно уменьшить влияние ионов водорода.
Если в анализируемом растворе помимо определяемых ионов присутствуют мешающие ионы, то есть ионы, к которым чувствителен данный электрод, то потенциал электрода зависит от концентрации как определяемых, так и мешающих ионов. Например, нитрат-селективный электрод характеризуется низкой селективностью к галогенид-ионам. Мешающее влияние последних устраняют, осаждая их сульфатом серебра. В паспортных данных для большинства наиболее часто применяемых электродов приведены перечень мешающих ионов и коэффициенты селективности электродов по отношению к этим ионам.
С помощью прямых потенциометрических измерений нельзя определить также ионы, которые взаимодействуют с другими содержащимися в растворе ионами или адсорбируются на поверхности твердой фазы, присутствующей в растворе. В этом случае необходимо разрушить соединение, в котором связан определяемый ион, или растворить осадок, на котором он сорбирован. Демаскировать определяемые ионы можно прибавлением реагента, который взаимодействует со связывающими ионами. Так, в растворе, содержащем ионы Fe2+, результаты определения фторид-ионов окажутся заниженными вследствие связывания ионов F- в FeF2. Для демаскирования фторид-ионов к раствору добавляют 1, 2-диаминоциклогексантетраацетат натрия, который образует комплексы с Fe2+, освобождая фторид-ионы. Сформулировать общие правила, которыми можно было бы руководствоваться при устранении мешающего действия различных веществ, довольно сложная задача.
В каждом конкретном случае важно правильно выбрать также электролит для заполнения жидкостного соединения (солевого мостика) Универсального электролита, одинаково пригодного для всех аналитических определений, не существует. При выборе электролита необходимо руководствоваться следующими положениями:
- ионная сила раствора в солевом мостике должна быть значительно больше, чем ионная сила стандартных и анализируемых растворов;
- электролит должен удовлетворять условиям эквитранспорта, то есть скорости диффузии положительных и отрицательных ионов должны быть по возможности одинаковыми;
- ионы, входящие в состав электролита, не должны взаимодействовать с компонентами анализируемого раствора;
- не рекомендуется использовать для заполнения солевого тика такие растворы, в состав которых входит определяемый ион или ион, существенным образом влияющий на отклик индикаторного электрода, что особенно важно при определении микроколичеств вещества в небольшом объеме анализируемого раствора.
Перед началом работы индикаторный электрод следует активировать путем повторяемых несколько раз измерений в стандартных растворах с различной концентрацией определяемого вещества. Такие измерения повторяют до тех пор, пока отклик электрода не станет быстрым и воспроизводимым. Обычно потенциал электрода считают воспроизводимым, если его величина изменяется не более чем на ± 0, 1 мВ за 1 минуту (для более точных измерений - не более, чем на ± 0, 1 мВ за 2 минуты). Иногда время отклика оп-ределяют как время, необходимое для достижения 95% от величины показаний электрода в стационарном режиме.
При измерениях, не требующих высокой точности, достаточно градуировать электрод по одному или двум стандартным растворам, а для точных измерений градуировочный график необходимо строить по большему числу точек. Для этого готовят серию стандартных растворов с известными концентрациями определяемого вещества, опускают в каждый из растворов соответствующие индикаторный электрод и электрод сравнения и измеряют э. д. с. На полулогарифмической диаграммной бумаге строят зависимость показаний прибора (в мВ) от концентрации для серии стандартных растворов (рисунок 1 ).
Поскольку сигнал электрода связан с активностью, а не с концентрацией определяемого вещества, то в области высоких концентраций может наблюдаться отклонение градуировочного графика от прямолинейной зависимости. Нелинейность графика обусловлена уменьшением коэффициента активности определяемого иона по мере увеличения концентрации электролита. Добавление избытка инертной соли позволяет поддерживать ионную силу раствора постоянной. В этом случае электродная функция линейна в широком диапазоне концентраций [6].

Рисунок 1 - Зависимость потенциала электрода от концентрации стандартного раствора
Современные аналитические приборы способны хранить в памяти значения откликов, полученных для градуировочных растворов, сравнивая их со значениями для анализируемого раствора и рассчитывая концентрацию определяемого вещества по заданному алгоритму с выдачей ее на дисплей прибора. Такие приборы позволяют своевременно выявлять и устранять грубые погрешности, корректировать наклон градуировочной зависимости и временной дрейф потенциала индикаторного электрода, а также учитывать поправку холостого опыта. В более сложных измерениях, требующих повышенной точности, используются компьютеры, контролирующие алгоритм собственно аналитического определения и обрабатывающие полученные данные. На экране дисплея можно отобразить также функцию погрешностей.
Однако получить действительно хорошие результаты измерений далеко не всегда просто. Для получения точных и воспроизводимых результатов первостепенное значение имеет постоянство условий на границе раздела электрод/анализируемый раствор. В каждом конкретном случае оно достигается путем выбора оптимальных условий эксперимента на всех стадиях, начиная от подготовки пробы и кончая обработкой полученных данных По этой причине методы потенциометрии часто проигрывают при сравнении со спектральными методами. Тем не менее они широко применяются при решении многих проблем аналитической химии, как определение фторидов в природных водах, определение ионов в биологических жидкостях и др. Поэтому химик-аналитик должен уметь применять эти методы.
1. 1. 2 Потенциометрическое титрование
Ещё более широкое применение в аналитической химии нашло потенциометрическое титрование, которое заключается в потенциометрическом наблюдении за ходом химической реакции. Потенциометрическое титрование можно охарактеризовать как титрование, при котором изменение э. д. с. гальванического элемента записывают в виде функции добавленного титранта [6].
Для успешного проведения титрования необходимо выполнение по крайней мере двух условий. Во-первых, в условиях проведения титрования химическая реакция между титрантом и изучаемым веществом должна быть стехиометрической. Во-вторых, при проведении титрования необходимо точно определять конечную точку титрования, которая должна быть достаточно близка к точке эквивалентности стехиометрической реакции титрования. Проведение количественных определений методом титрования возможно только при одновременном выполнении этих двух условий [1].
Для потенциометрического титрования собирают цепь из индикаторного электрода и электрода сравнения. Индикаторный электрод выбирают в зависимости от реакции, которая лежит в основе титрования. Для титрования по методу нейтрализации используют, как правило, стеклянный электрод, однако возможны и другие (водородный, хингидронный, сурьмяный). Для титрования по методу окисления-восстановления используют редокс-электроды, по методу осаждения и комплексообразования также редокс-электроды или электроды, соответствующие типу реакции. В качестве электродов сравнения используют хлорсеребряный или каломельный электроды [4].
Потенциометрическое титрование объединяет способы определения конечной точки титрования (KTT), основанные на зависимости потенциала индикаторного электрода от объема добавленного титранта. По сравнению с прямыми измерениями полученные при потенциометрическом титровании данные более точно и правильно характеризуют концентрацию определяемого вещества, поскольку не зависят от его активности. Кроме того, в методах потенциометрического титрования к электродам предъявляются менее жесткие требования в отношении стабильности потенциала и крутизны наклона электродной функции. Электроды, непригодные для прямых потенциометрических измерений, могут отвечать требованиям потенциометрического титрования. Наконец, методы потенциометрического титрования позволяют находить концентрацию анализируемого компонента даже в присутствии мешающих ионов, если титрант селективно взаимодействует с определяемым веществом [5].
Метод потенциометрического установления точки эквивалентности по своим возможностям превосходит титриметрические методы с применением цветных индикаторов. Он обладает большей точностью, чувствительностью, позволяет анализировать окрашенные и мутные растворы, допускает возможность дифференцированного определения веществ в одном растворе и позволяет автоматизировать процесс титрования. Как и в обычном титримет-рическом анализе, в потенциометрическом титровании используются реакции нейтрализации, осаждения, комплексообразования, окисления-восстановления. Требования к реакциям титрования те же, что и при химической индикации: высокая скорость прямой реакции; строгая стехиометрич-ность и практическая необратимость, отсутствие побочных реакций [7].
Для потенциометрической индикации точки эквивалентности необходимо, чтобы в области КТТ потенциал индикаторного электрода изменялся скачкообразно.
Обычно процесс титрования включает в себя измерение и запись потенциала индикаторного электрода после каждого прибавления к анализируемому раствору порции реагента. В начале титрования реагент добавляют большими порциями, а по мере приближения к точке эквивалентности (на это указывает большое изменение потенциала при добавлении реагента) объем добавляемого титранта уменьшают. Вблизи точки эквивалентности изменение э. д. с. становится наиболее заметным, так как именно в точке эквивалентности изменение концентрации раствора происходит с наибольшей скоростью.
Различают три основных способа потенциометрического титрования: S-, Т- и R-титрование. При проведении S-титрования применяют электрод, чувствительный к определяемому компоненту. По мере приближения к точке эквивалентности потенциал электрода изменяется в соответствии с уменьшением активности определяемых ионов. В точке эквивалентности активность ионов определяется константой равновесия соответствующей реакции (константой диссоциации образующегося соединения, константой нестойкости комплексного иона, произведением растворимости осадка т. д. ). При этом происходит скачкообразное изменение потенциала индикаторного электрода (рисунок 2 - а) [5].
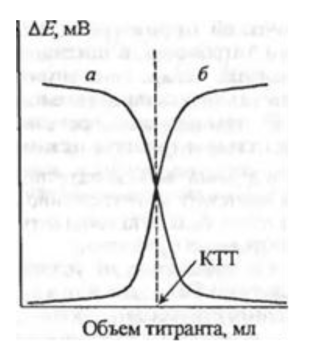
Рисунок 2 - Кривые потенциометрического титрования а - S-титрование; б - T-титрование
В случае T-титрования с помощью индикаторного электрода контролируют концентрацию титранта. Первоначально добавляемый титрант расходуется на связывание определяемых частиц, поэтому потенциал электрода вплоть до достижения точки эквивалентности изменяется незначительно. Появление избытка титранта после точки эквивалентности приводит к резкому увеличению его активности в анализируемом растворе и, следовательно, к изменению э. д. с. (рисунок 2 - б).
Метод R-титрования основан на использовании электрода, чувствительного к индикаторному иону. При этом индикаторный ион должен находиться в равновесии и с определяемым веществом, и с мигрантом, который может образовывать комплексы или малорастворимые соли с этими двумя веществами. Так, например, кальций определяют с помощью медь-селективного электрода: к анализируемому раствору добавляют ионы Сu2+, а затем титруют раствор ЭДТА. Поскольку титрант образует с Сu более прочный комплекс, чем с Са2+, то на кривой титрования наблюдаются две КТТ: первая соответствует меди, а вторая - кальцию. Если же индикаторный ион образует с титрантом более слабый комплекс, то к анализируемому раствору добавляют избыток титранта и оттитровывают непрореагировавший реагент раствором индикаторного иона. Разность между добавленным количеством титранта и его непрореагировавшим количеством позволяет вычислить концентрацию определяемого вещества. В качестве примера можно привести определение фосфат-ионов: к анализируемому раствору добавляют избыток нитрата лантана и оттитровывают непрореагировавшие ионы лантана раствором фторида, используя фторид-селективный электрод [5].
Точность результатов титрования в значительной степени зависит от надежности фиксирования точки эквивалентности и симметричности кривой титрования.
При потенциометрическом титровании с использованием неполяризо-ванных электродов измеряют равновесный потенциал электрода, находящегося в титруемом растворе. Путем подстановки потенциала электрода в уравнение Нернста можно рассчитать значения активности или концентрации потенциалопределяющего иона в любой точке кривой титрования вне зависимости от того, где находится эта точка - до или после точки эквивалентности или даже если она сама является этой точкой. При определенных условиях метод позволяет проводить титрование до теоретически рассчитанного значения потенциала электрода в точке эквивалентности или до потенциала, установленного при титровании стандартного раствора.
Скачок потенциала в области точки эквивалентности определяется разностью стандартных потенциалов соответствующих редокс-пар. Поскольку скачок потенциала составляет обычно несколько десятков или сотен милливольт, т е. Достаточно велик, то точку эквивалентности удается определить с необходимой точностью.
При рассмотрении индикаторных электродов, применяемых в потенциометрическом титровании, следует заметить, что только в окислительновосстановительных и кислотно-основных реакциях индикаторные электроды являются универсальными. Действительно, независимо от того, какие редокс-системы применяются при титровании, в качестве индикаторного электрода можно использовать один и тот же благородный металл (платина, золото и др. ) являющийся переносчиком электронов. То же самое можно сказать и об индикаторных электродах в методе кислотно-основного титрования: независимо от природы кислот, оснований и титрантов химическая реакция связана с изменением концентрации ионов водорода в растворе. Поэтому достаточно иметь любой индикаторный электрод, обратимый относительно Н+-ионов.
В осадительном и комплексонометрическом потенциометрическом титровании применяются более селективные индикаторные электроды. Это объясняется тем, что ионы, входящие в состав осадков или комплексов, имеют разную природу, а индикаторный электрод должен быть обратимым относительно хотя бы одного из них [5].
Точность метода составляет 0, 1 - 0, 5 %. Для нахождения точки эквивалентности используют 4 графических способа (рисунок 3):

Рисунок 3 - Графические способы определения точки эквивалентности
- по интегральной кривой титрования (а) - наиболее простой способ. Проводят 3 касательные (к двум пологим участкам кривой и к скачку), затем полученный между двумя точками пересечения отрезок делят пополам и опускают перпендикуляр на ось абсцисс.
- по дифференциальной кривой титрования (б) - более точный способ.
- по второй производной (в) - используется для более точного определения точки эквивалентности в случае ассиметричного или малого скачка. Соединяют концы ветвей графика. На пересечении этой прямой с осью абсцисс находят точку эквивалентности.
- по кривой в координатах AV/AE - V(R) (метод Грана) (г) - используется для разбавленных растворов [8].
1. 2 Электроды в потенциометрическом анализе
В последние годы большое внимание уделяется разработке первичных измерительных преобразователей (датчиков, или сенсоров) различных физических величин: температуры, давления, ускорения, концентраций ионов в жидкостях, химического состава газовых сред и др. Эти сенсоры находят все большее применение в химической и электронной отраслях промышленности, машиностроении, авиационной и автомобильной технике, космонавтике, при добыче и транспортировке нефте- и газопродуктов, в медицине.
Сенсоры, как правило, должны избирательно (селективно) воспринимать физическую величину, подлежащую измерению, и преобразовывать ее в другую величину, удобную для сравнения с мерой и для последующей об -работки измерительного сигнала [9].
Вряд ли можно какой-либо электрод назвать универсальным, то есть имеющим все необходимые свойства для использования в электрохимическом анализе. Можно говорить лишь о материалах и способах, которые применяются для изготовления электродов. Многие из них были созданы специально для того или иного метода электрохимического анализа (стеклянные, ионоселективные, ферментные электроды и др. ) [5].
В настоящее время широкое применение нашли ионоселективные электроды мембранного типа, в которых перенос электрического тока осуществляется ионами. Тем не менее используются также и классические металлические электроды с электронным, а не ионным переносом; в частности, они входят в качестве составной части в электроды сравнения, которые сами по себе заслуживают особого внимания.
Независимо от природы электрода возникающий на нем потенциал подчиняется уравнению Нернста:
![]()
где k/z - характеристика электрода, называемая крутизной электродной функции, теоретически равная величине (RT/zF) ln (R - газовая постоянная, Т - абсолютная температура, F - число Фарадея, z - заряд иона) [10].
Любая электрохимическая ячейка или электрохимический прибор должны иметь, по меньшей мере, два электрода и один электролит. Под электродом понимают границу раздела, на которой электронный (направленное движение электронов) механизм переноса заряда меняется на ионный (направленное движение ионов) и наоборот, а под электролитом понимают среду, в которой осуществляется перенос заряда направленным движением ионов. В менее формальном смысле термин электрод применяют для обозначения электронного проводника, а термин электролит — для обозначения ионного проводника в электрохимической ячейке. Простейшую электрохимическую ячейку можно представить так, как это сделано на рисунке 4.
В любой электрохимической ячейке должно быть минимум два физических электрода. Однако в ячейке, применяемой для электрохимических измерений, всегда присутствуют три электродные функции. Это положение становится более очевидным при проведении неравновесных измерений, когда от ячейки отводят значительный ток, поскольку в такой ситуации эти три электродные функции осуществляются с помощью трех различных физических электродов; однако это положение остается справедливым и для равновесных измерений, даже если их проводят с помощью двух физических электродов [2].
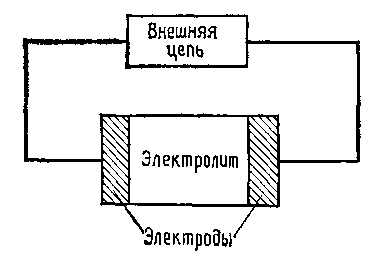
Рисунок 4 - Схема электрохимической ячейки [2]
Первый из трех электродов в соответствии с выполняемой им функцией называют индикаторным. Некоторые авторы называют его также испытательным электродом или рабочим электродом. Это электрод, на котором происходят исследуемые электрохимические процессы. При помощи этого электрода проводятся электрохимические измерения. Он может быть изготовлен из инертного или какого-либо другого материала.
Второй функциональный электрод - это электрод сравнения, или непо-ляризованный, или неполяризующийся электрод. Он обладает постоянным потенциалом, достаточно стабильным для того, чтобы использовать его в качестве эталона сравнения, относительно которого измеряют потенциалы других электродов ячейки. Под словами «достаточно постоянный потенциал» мы понимаем следующее: изменение потенциала под действием тока, времени или других переменных величин необязательно должно быть равно нулю, но должно быть сравнительно небольшим, и, кроме того, проводимые измерения не должны вызывать необратимого изменения величины потенциала электрода. Этот электрод не следует изготавливать из инертного материала.
Третьим функциональным электродом является противоэлектрод, известный также как вспомогательный электрод. Он служит источником электронов или выполняет роль стока электронов и тем самым обеспечивает возможность протекания тока через ячейку. Как правило, ни его ток, ни его потенциал не измеряются. Его обычно изготавливают из инертного материала.
Индикаторный электрод в связи с выполняемой им функцией нельзя объединить ни с одним из других электродов, и, следовательно, он всегда существует как отдельный физический электрод. Другие две электродные функции можно иногда объединить для того, чтобы в системе присутствовало только два физических электрода. Единственное преимущество такого объединения — более удобная геометрия ячейки и внешнего оборудования к ней. В то же время такое объединение нежелательно из-за несовместимости функций электрода сравнения и противоэлектрода, состоящей в том, что потенциал, стабильность которого необходима для электрода сравнения, может необратимо изменяться под действием тока, требуемого для работы противо-электрода. Тем не менее в электрохимических методах анализа можно использовать ячейку с двумя электродами, если точно известно, что величина тока, протекающего через ячейку, достаточно мала, чтобы эффект необратимого изменения потенциала, называемого поляризацией электрода сравнения, имел незначительную величину. Например, в классической водной полярографии применяют такие двухэлектродные ячейки с насыщенным каломельным электродом в качестве совмещенного электрода сравнения и вспомогательного электрода. Замена трехэлектродной ячейки на двухэлектродную никогда не приводит к улучшению результатов измерений; при этом можно получить ячейку более простую по конструкции, но она никогда не будет обладать лучшими параметрами [2].
Из-за большого различия в методах электрохимического анализа рекомендовать какую-то одну универсальную конструкцию ячейки не представляется возможным. Обычно электрохимические ячейки изготавливают из какого-либо твердого химически стойкого материала, например термостойкого стекла или кварца. В последнее время для этих целей используют тефлон и другие полимерные материалы. Однако в присутствии неводных растворителей органические вещества из полимеров могут переходить в анализируемый раствор, снижая тем самым чувствительность измерений. Тефлон выгодно отличается от других материалов не только химической инертностью, но и тем, что на нем практически не адсорбируются многие ионы металлов и органические соединения, что важно при их определении на уровне следовых количеств [5].
Классификация ионселективных электродов основана на различии селективных химических реакций, которые приводят к образованию межфаз-ного потенциала. Специфическое распознавание потенциометрическим химическим сенсором достигается благодаря химической реакции на поверхности сенсора. Таким образом, поверхность электрода должна содержать реагент, который химически и обратимо взаимодействует с аналитом. Это достигается благодаря использованию ионселективных мембран, которые представляют собой поверхность сенсора. Раньше считалось, что ионселективные электроды, в которых использованы такие мембраны, являются «специфическими» для конкретного иона. Теперь мы знаем, что на химическое равновесие на поверхности сенсора могут влиять и другие ионы, поэтому более правильно при описании этих сенсоров использовать термин «селективный», а не «специфический».
В потенциометрических химических сенсорах используются четыре типа мембран [11].
- Стеклянные мембраны. Селективны по отношению к таким ионам, как Н+, Na+ и NH4+.
- Мембраны из плохо растворимых неорганических солей. К мембранам этого типа относятся монокристаллические неорганической соли, например LaF3, или диски из спрессованного порошка неорганической соли или смеси солей, например, Ag2S/AgCl. Эти мембраны селективны по отношению к таким ионам, как F-, S2- и С1-.
- Полимерные мембраны с иммобилизованным ионофором. В этих мембранах ионселективные комплексообразуюшие соединения или ионо-обменники иммобилизованы в полимерной матрице, например, в поливинилхлоридной.
- Мембраны с иммобилизованными в геле или химически связанными с гелем ферментами. В таких мембранах используются высокоспецифичные реакции, катализируемые ферментами. Фермент содержится внутри матрицы или химически прививается на твердой поверхности.
1. 2. 1 Ионоселективные электроды с твёрдыми мембранами
В зависимости от применяемого электродно-активного материала ионоселективные электроды можно разделить на электроды с твердой и жидкостной мембраной.
Электроды с твердой мембраной можно в свою очередь подразделить на стеклянные электроды, электроды с кристаллической мембраной и гомогенные или гетерогенные осадочные мембранные электроды. Вследствие сравнительно плохой электропроводности материала электродов, как правило, применяют мембраны небольшой толщины и поэтому говорят о мем
бранных электродах. Слово «мембрана» не должно приводить к ошибочному представлению о том, будто речь может идти о толщине биологических мембран. В зависимости от удельного сопротивления материала можно вычислить толщину мембран для разного типа электродов; так, стеклянные ~ 0, 1 мм, из органических материалов примерно в пределах 1 — 5 мм, монокри-сталлические и осадочные мембраны >3 мм.
Одна из проблем ионоселективных электродов заключается в измерении потенциала соответствующей фазы электродного материала, не оказы-ваая на него влияния. Не всегда можно конец измерительного проводника поместить в активный материал, специфически обменивающийся ионами с анализируемым раствором. Именно для этого создают добавочную границу раздела фаз с дополнительным изменением напряжения тока измерительного контура. На этой границе идет обратимая реакция, в результате чего возникает постоянный гальвани-потенциал. Место контакта должно представлять собой неполяризуемый электрод, хотя это требование трудновыполнимо. В каждом случае на измеряемой стороне мембраны протекает в одном направлении обратимая и неполяризуемая электродная реакция с определяемым ионом (в противном случае работа электрода не подчинялась бы уравнению Нернста). На другой стороне ионселективной мембраны идет такая же реакция, но при постоянной активности соответствующего определяемого иона.
Поэтому проблему передачи потенциала ионселективной фазы без оказания на него влияния можно решить, если мириться с добавочным постоянным гальвани-потенциалом на новой границе раздела фаз.
Между тем измерение потенциала на границе раздела фаз ионселек-тивной мембраны, противоположной измеряемой стороне, представляет собой большую проблему. Каким образом осуществляется вывод потенциала внутреннего раствора к входным клеммам вольтметра? Нет иного решения, как признать наличие добавочной разности потенциалов при введении дополнительной границы раздела фаз. При этом еще раз необходимо отметить то, что гальвани-потенциал остается постоянным. Это можно сделать, использовав сравнительный полуэлемент. Необходимо следить только за тем, чтобы во внутреннем растворе активность ионов, участвующих в реакциях на границе раздела фаз, оставалась постоянной. Если во внешней и внутренней цепи сравнения применяют одинаковые сравнительные полуэлементы, то речь идет о симметричных измерительных цепях. Эти цепи предпочтительны вследствие их компенсирующего действия. На рисунке 5 приведены конструкции ионоселективных электродов [12].
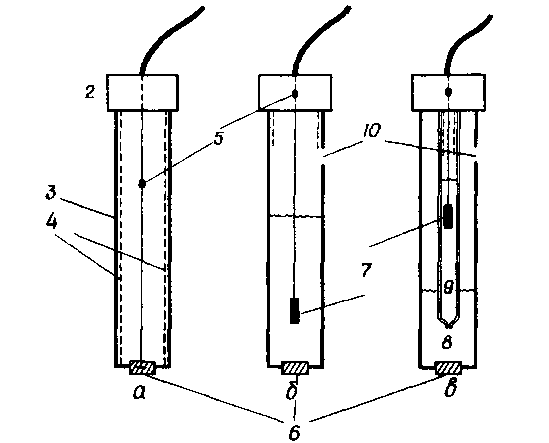
а - Прямой контакт электронного проводника с активной фазой, б - Промежуточное включение внутреннего раствора и вывода при помощи обратимого полуэлемента. в - Промежуточное включение внутреннего раствора и вывода через соответствующий электрод сравнения с солевым мостиком.
1 - кабель; 2 - крышка; 3 - корпус электрода (пластмасса, стекло); 4 - экран; 5 - спай; 6 - ионоселектнвная фаза; 7 - сравнительный полуэлемент (в большинстве случаев Ag/AgCl); 8 - внутренний раствор; 9 - солевой мосток.
Рисунок 5 - Конструкции ионоселективных электродов с твердой активной фазой [12]
В некоторых случаях (например, у электродов с твердыми мембранами) внутренний контакт к измерительному прибору, посредством которого осуществляется переход от ионного к электронному проводнику, удается сделать неполяризуемым (обратимые окислительно-восстановительные реакции). При использовании такого контакта «твердое - твердое» нет необходимости применять внутренний раствор с постоянной ионной активностью. Поэтому такие электроды называют бесконтактными. С другой стороны, при использовании электродов такой конструкции нужно учитывать, что э. д. с. электрода сравнения нельзя скомпенсировать. Температурная характеристика в этом случае может быть совсем иной, чем у симметричных цепей; кроме того, поляризуемость у них больше [12].
Обычно мембранные электроды используют вместе с отдельным электродом сравнения, который помещают в испытуемый раствор. Твёрдую мембрану изготавливают либо из монокристаллического вещества (например, LaF3 для фторидселективного электрода), либо из поликристаллического порошкообразного вещества (например, AgS для сульфидселективного электрода) [13] или керамических материалов, получаемых спеканием и прессованием при высокой температуре [14]. К электродам с гомогенными мембранами относится также стеклянный электрод.
Честь первооткрывателя ИСЭ с мембранами, представляющими собой осадки умеренно растворимых солей, внедренных в инертную матрицу, принадлежит Пунгору. Гомогенные мембраны имеют определенные преимущества над гетерогенными мембранами с точки зрения воспроизводимости отклика соответствующего электрода, однако для изготовления гомогенных мембран требуются специальные методические приемы, в то время как осадочные мембраны могут быть получены в сравнительно простых лабораторных условиях. Для изготовления электродов с матрицей из силиконовой резины Пунгор и сотрудники использовали следующую методику: смесь осадка соответствующей соли и полисилоксана гомогенизируют и добавляют сшивающий реагент (производное силана) и катализатор с таким расчетом, чтобы смесь содержала около 50% соли. Требуемую форму мембраны получают ка-ландрованием. Качество мембраны зависит от степени сшивания матрицы, поскольку число поперечных связей определяет распределение частиц осадка в мембранной фазе. Бухапаи и Сиго рекомендуют смешивать силиконовую резину с порошкообразными галогеиидами серебра и прессовать смесь между полиэтиленовой пластинкой и поливинилхлоридной пленкой. Другие методики изготовления гетерогенных мембран включают осаждение галогени-дов серебра в термопластовую или полиэтиленовую матрицу. В последнем случае мембраны следует прессовать при температуре от 100 до 130°С и давлении 1х107 - 3х107 Па. Подходящим материалом для матрицы мембраны является также дентакрил [14, 15].
Кристаллические мембраны отличаются очень высокой селективностью, превышающей селективность жидкостных электродов (с ионообменными веществами) на несколько порядков. Это связано с тем, что селективность у твердых кристаллических мембранных электродов достигается за счет вакансионного механизма переноса заряда, при котором вакансии заполняются только определенным подвижным ионом (например, Ag+), так как форма, размер, распределение заряда вакансии соответствуют только определенному подвижному иону.
Теория функционирования твердых кристаллических электродов разрабатывается и совершенствуется уже много лет школой Бака, который детально рассматривает особенности твердых электродных мембран (главным образом, в кинетическом аспекте). Аналогия между мембранными электродами из AgX (где X - С1-, Вr-, I-) и электродами второго рода вполне очевидна, но кинетика достижения равновесия для них может существенно различаться [16].
Поликристаллическая мембрана свинцового твердого электрода получена из смеси PbS и Ag2S путем прессования [17]. Фирма Orion выпускает PbS-электроды со следующими характеристиками: концентрационный интервал 100 - 10-7 М, электрическое сопротивление 1 МОм, температурный интервал 0 - 80 °С, допустимое содержание ионов Ar+, Hg2+, Cu2+- не более 10-7 М. Высокое содержание ионов Cd2+ и Fe3+ приводит к нарушению Pb2+-функции электрода.
Электроды с поликристаллическими мембранами описаны также в работах А. В. Гордиевского (мембраны с PbS, введенным в силиконовую матрицу, также функционируют как Pb -электроды) [16].
Время отклика свинцовых гетерогенных поликристаллических электродов менее 2 мин; интервал pH, в котором их можно применять, 2, 8 - 7, 0. Нернстовская зависимость потенциала от активности ионов Pb2+ выполняется во всем интервале его функционирования. Предложен также способ термической обработки смеси PbS - Ag2S с полимером для формирования мембраны свинцового электрода, который показал удовлетворительные характеристики.
В работе [18] изучено влияние условий прессования смеси PbS и Ag2S на электродные свойства мембраны. Наилучшим оказалось давление 1 кПа (при 350—500°С в течение 3 ч. Такой электрод обладает полной электродной функцией в концентрационном интервале ионов Pb2+ 10-1 - 10-6 М.
Отметим, что разработан также селектрод [19], обладающий Pb2 функцией в интервале концентраций 10-2 - 10-11 М Pb2+, что позволило использовать этот селектрод при потенциометрическом титровании с ЭДТА и нитрилотриуксусной кислтой [20].
Для свинцовых электродов применяли также селениды и теллуриды свинца. В работе японских учёных [21] описываются ионоселективные электроды, состоящие из смеси селенида или туллурида свинца с сульфидом серебра, спечённые в мембраны при температуре 600°С. Полученные таким образом мембраны давали линейный отклик на ионы свинца в диапазоне кон центраций 1x10-1 до 1x10-7 с наклоном 29, 5 мВ/pPb (рисунок 6). По сравнению с сульфидсвинцовыми мембранами мембраны из селенида и теллурида оказались более устойчивыми в сильнокислых средах. Нернстовская зависимость наблюдалась в интервале температур 0 - 95 °С.
Чувствительность к ионам свинца уменьшается в ряду PbS » PbSe >PbТе, а чувствительность к H+ соответствует иному ряду: PbS <PbSe « PbТе [22].
Халькогенидные электроды малопригодны в прямых измерениях, но их используют при потенциометрическом титровании свинца. Ионами, влияющими на потенциал свинцового сульфидного (халькогенидного) электрода гомогенного и гетерогенного типа, являются Ag2+, Hg2+, Cu2+, Fe3+, S2- и I-. В прямых потенцио- и титриметрических определениях эти ионы должны практически отсутствовать [16]. Ионы Al3+, Zn2+, Cd2+, Co2+, Ni2+, Mg2+ и нитрат-ион на точность определений не влияют [21].
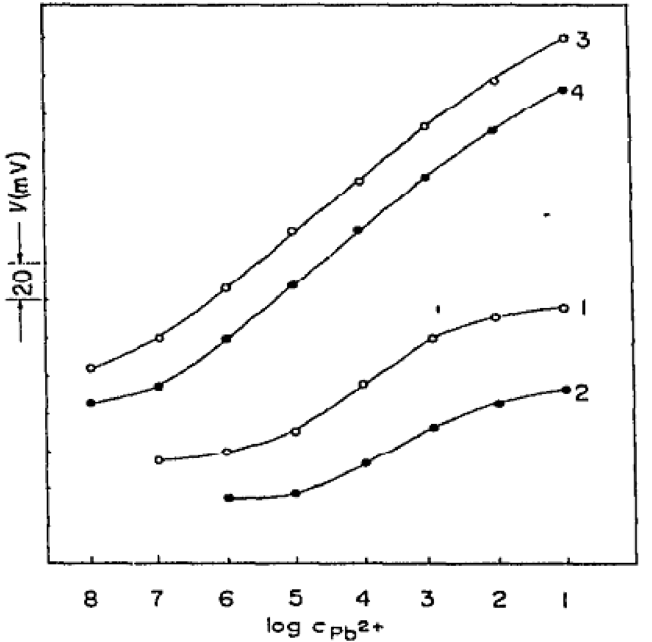
1 - спрессованный PbSe; 2 - спрессованный PbTe; 3 - спечённый PbSe; 4 -спечённый PbTe
Рисунок 6 - Потенциальные кривые для свинецселективных мембранных электродов [21]
Ионоселективный электрод с мембраной из сульфида свинца импре-гнированного в силиконовую резину также был изучен [17]. Показано, что ионы: K+, Na+, Mg2+, Ca2+, Al3+, Cr3+, Zn2+, Fe2+, Co2+, Ni2+, NH4+, NO3-, ClO4- и
2+CH3COO- не мешают определению Pb -иона, если их соотношение с концентрацией определяемого иона не превышает 1x103. Более чем 1x10-3 MCl-,
Br-, I-, SO4 2- мешают определению из-за образования нерастворимых солей свинца, а Cu2+, Ag+, Hg2+ не могут сосуществовать из-за того, что они осаждаются на поверхности мембраны, так как растворимость их сульфидов меньше, чем растворимость сульфида свинца.
Подобный PbS-электрод перед использованием должен не менее семи
дней вымачиваться в 1x10-2 М растворе нитрата свинца, а его рабочая область pH ограничена интервалом от 2, 8 до 7. Зависимость E =ƒ(pPb2+) для подобного электрода, измеренная на водных растворах нитрата свинца представлена на рисунке 7 [17].
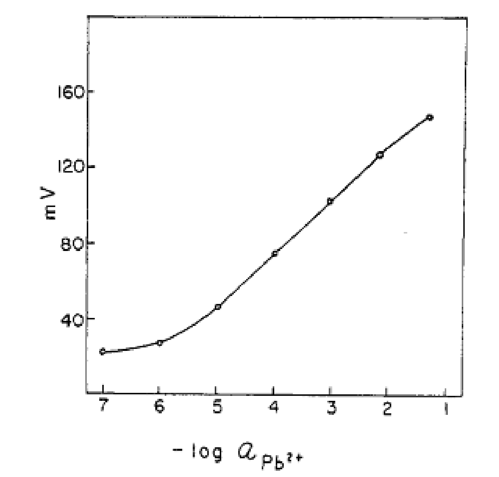
Рисунок 7 - Потенциальная кривая для свинецселективного мембранного электрода из силиконовой резины, пропитанной PbS [17]
Электрод фирмы Orion, состоящий из смеси сульфидов свинца и серебра использовали при прямом титровании сульфатов в 50 % растворе n-диоксана. В растворах, в которых пытаются оценить микроколичества сульфата титрованием с раствором перхлората свинца, должен отсутствовать PbSO4 и фосфаты, а C1- и NO3- мешают титрованию, если присутствуют в 100-кратном избытке.
С помощью РЬ2+-селективных электродов на основе халькогенидов свинца определяли содержание серы в органических соединениях в 60 % n-диоксане, полумикроколичества оксалата в 40 % n-диоксане, а также микроколичества ортофосфата методом прямого потенциометрического титрования. В последнем случае значение pH растворов поддерживали на уровне 8, 25 - 8, 75 с помощью буферных систем. Присутствие же NO3- и SO4 2- лишь в небольшой степени мешало функционированию электрода в соответствии с уравнением Нернста. То же относится к Cl- и F-, хотя их наличие приводило к завышению определяемых количеств фосфатов [20]. Аналогичными заводскими электродами определялось содержание сульфатов методом осадительного титрования в среде метанола, изопропанола и смеси с n-диоксаном [23].
РЬ2+-селективный электрод применяли в качестве диагностического средства для установления факта свинцового отравления у детей, для определения SO2 в топочных газах, для измерения констант ассоциации PbSO4 и нахождения суьфатов в минеральной и морской воде, определяли концентрацию свинца в крови, слюне и моче человека [20, 24].
2 Экспериментальные методы исследования ионоселективных и индикаторных свойств халькогенидов свинца
2. 1 Физико-химические свойства халькогенидов свинца
Халькогениды свинца относятся к бертоллидам - классу соединений нестехиометрического состава. Склонность к образованию таких соединений повышается от серы к теллуру. Например, в сульфиде свинца отклонение от стехиометрии лежит в пределах 0, 1 %, но оно вызывает изменение твёрдости, отражения и проводимости [1].
Халькогениды свинца (сульфид, селенид и теллурид) в стандартных условиях представляют собой серебристо-серые кристаллы кубической син-гонии (рисунок X) типа NaCl (z = 4, пространственная группа Fm3m). Однако, для сульфида свинца при повышении давления до 2, 4—4, 2 МПа устойчивой становится ромбическая сингония (типа SnS, пространственная группа Pcmn) [2].
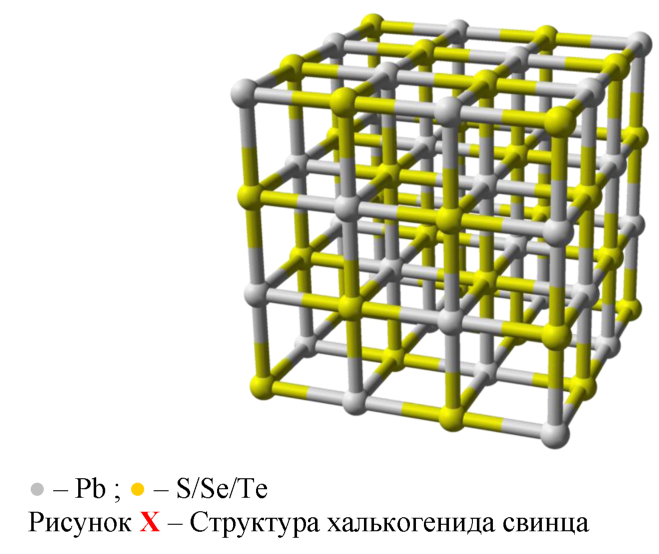
Несмотря на совпадение структуры кристаллической решётки халько-генидов свинца с типом, свойственным ионным кристаллам, химическая связь носит преимущественно ковалентный характер. Координация атомов кристаллической решётки объясняется с физико-химических позиций p -гибридизацией (в отличии от sp -гибридизации, типичной для алмазоподобных полупроводников). Все PbX (X = S, Se, Te) имеют зонную структуру.
В таблице 1 приведены некоторые физические характеристики халькогенидов свинца [2, 3].
|
Халькогенид |
Температура плавления, К |
Плотность,
г/см3 |
Характер проводимости |
Ширина запрещённой зоны (Eq(300К) эВ |
|
PbS |
1351 |
7, 61 |
p-типа |
0, 41 |
|
PbSe |
1338 |
8, 26 |
p-типа |
0, 28 |
|
PbTe |
1193 |
8, 24 |
p-типа |
0, 32 |
2. 1. 1 Сульфид свинца
В системе Pb-S имеется одно соединение PbS. Диаграмма состояния этой системы приведена на рисунке 2. В природе PbS встречается в виде минерала галенита - одного из наиболее распространённых свинцовых минералов [4].
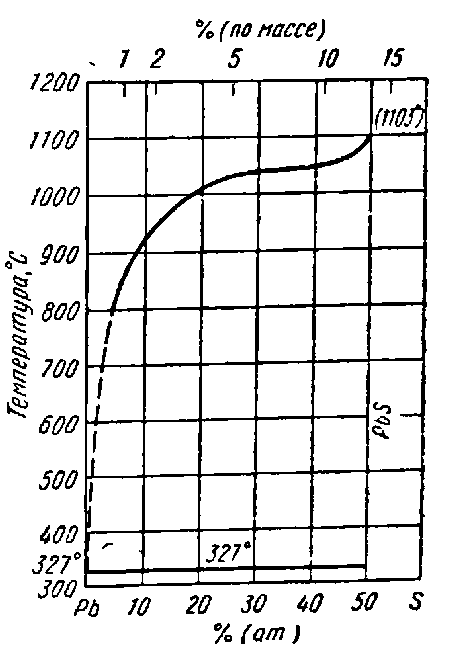
Рисунок 2 - Диаграмма состояния системы Pb-S
Сульфид свинца представляет собой кристаллическое соединение с окраской от сине-серой до серебристо-серой.
При осаждении растворов солей свинца действием сероводорода в присутствии кислоты образуются аморфные или частично кристаллические осадки PbS. Однородные кристаллические осадки сульфида свинца получают взаимодействием раствора плюмбита натрия с тиомочевиной при нагревании. Кроме того, сульфид свинца может быть получен из элементов постепенным, с выдержками, нагреванием ампулы со смесью веществ [4].
Сульфид свинца имеет решётку типа NaCl с параметром а = 5, 9362 А. Основой структуры является кубическая плотнейшая упаковка анионов в параллельных плоскостях и как катион, так и анион имеют правильную октаэдрическую шестерную координацию [1]. Область гомогенности его очень узка: ±3x10-4 атом/моль. Температура плавления сульфида свинца, по данным различных авторов, лежит в пределах 1119 ± 16°С. Другие физические и физико-химические свойства PbS представлены в таблицах 1 и 2 [2, 3].
PbS трудно растворим в воде и имеет слабо щелочную реакцию. В 100 мл воды при комнатной температуре растворяется 3x10-5 г сульфида (-lg(KS) = 28, 1). Сульфид свинца растворяется в разбавленной соляной кислоте, в концентрированной HCl растворение идёт очень медленно. Разбавленная азотная кислота разлагает PbS даже на холоду, а растворы едких щелочей разлагают сульфид свинца лишь частично [4].
Таблица 2 - Физико-химические характеристики халькогенидов свинца
|
Характеристика |
Халькогенид свинца |
||
|
PbS |
PbSe |
PbTe |
|
|
Параметр кристаллической решётки, нм |
0, 593 |
0, 613 |
0, 646 |
|
C0p, Дж/(МольxК) |
49, 50 |
50, 21 |
50, 54 |
|
∆Н0fe, кДж/моль |
36, 5 |
35, 6 |
41, 1 |
|
∆Н0lad, кДж/моль |
-99, 6 |
-99, 2 |
-68, 6 |
|
S0298, Дж/(МольxК) |
91, 2 |
102, 5 |
110, 0 |
|
Теплопроводность, Вт/(мxК) |
2, 5 |
1, 6 |
2, 0 |
|
Подвижность электронов при 300 К, см2/(Вxс) |
6x102 |
1x103 |
2x103 |
|
Подвижность дырок при 300 К, см2/(Вxс) |
6x102 |
1x103 |
8x102 |
|
Концентрация собственных носителей заряда при 300 К, см'3 |
2x1015 |
3x1016 |
1, 5x1016 |
|
Показатель преломления n (λ = 3 мкм, T = 300 К) |
4, 10 |
4, 59 |
5, 35 |
2. 1. 2 Селенид свинца
Диаграмма состояния Pb-Se исследовалась неоднократно (рисунок 3). Методами термического, рентгенографического, микроструктурного анализа и ряда других физических способов установлено, что PbSe (27, 59 вес. % Se) является единственным соединением [5].

Рисунок 3 - Диаграмма состояния системы Pb-Se
PbSe — свинцово-серое, слегка голубоватое вещество, в котором четыре валентных электрона свинца распределены по орбитам 6s2p2, а шесть валентных электронов селена соответственно по орбитам 4s2p4. PbSe нерастворим в воде (-lg(Ks) = 36, 2) и многих других растворителях. Селенид свинца имеет кубическую решетку типа NaCl с параметром а = 6, 122-7-6, 147 А.
Энергия решетки PbSe равна 735 ккал/моль. Плотность PbSe колеблется в интервале 8, 01—8, 29 г/см; разность электроотрицательностей 0, 7; ионизационный потенциал 15, 0 эВ, а показатель преломления - 4, 54. Энергия диссоциации PbSe 81, 0 ккал/моль. Диэлектрическая постоянная для селенида свинца определена равной 20. PbSe — изоморфен с галенитом; размеры ячейки возрастают от галенита к PbSe (клаусталлит) по мере увеличения концентрации селена [5].
Наиболее простой способ получения селенида свинца состоит в сплавлении элементарных селена и свинца в стехиометрических количествах. Кроме того, селенид свинца можно получить при взаимодействии солей свинца с селеноводородом или селеномочевиной в присутствии гидразина, как основания.
Селенид свинца может иметь как дырочную, так и электронную проводимость. Удельное сопротивление колеблется в интервале 5x10-2-5x10-3 омxсм. Подвижность электронов при комнатной температуре ~ 1175; дырок ~ 500 см /вxсек, а время жизни носителей 0, 6 мксек [5].
2. 1. 3 Теллурид свинца
На диаграмме состояния Pb-Te отмечается одно химическое соединение - PbTe, содержащее 38 вес. % Te (рисунок 4). PbTe, образующийся в результате взаимодействия свинца с теллуром, плавится с открытым максимумом при 917°С и имеет интервал гомогенности, пределы которого изменяются в зависимости от температуры и скорости охлаждения [6].

Рисунок 4 - Диаграмма состояния системы Pb-Te
Теллурид свинца представляет собой серебристо-серое твёрдое вещество, кристаллизующееся в кубической сингонии, структурного типа NaCl с параметром решётки а = 6, 439 А и координационным числом равным четырём.
Теплота образования теллурида свинца по реакции Pb + Te = PbTe составляет 81 ккал/моль. Разность электроотрицательностей Pb и Te равна 0, 5, доля ионной связи составляет 9 %. Теллурид свинца и сплавы системы свинец-теллур могут быть приготовлены сплавлением исходных компонентов в эвакуированных кварцевых сосудах [6].
В природе теллурид свинца встречается в виде минерала алтаита. Минерал редко встречается в виде кубов и октаэдров, обычно он представлен сплошными массами.
Теллуристый свинец - кристаллическое вещество, в котором преобладают атомо-ковалентные силы связи. Коэффициент теплопроводности PbTe при 25°С равен 5, 25х10-3 кал/смxсекxград. Теллурид свинца с концентрацией носителей заряда 10 - 10 см-3 имеет удельную электропроводность в пределах 60-1800 ом-1xcм-1 при 25°С [6].
2. 2 Синтез и идентификация халькогенидов свинца
2. 3 Растворы, установка и методика измерений
Для проведения исследований были приготовлены и применялись следующие растворы:
- раствор нитрата свинца (II) 0, 1 - 1х10-9 моль/л, приготовленный растворением точной навески (чда) в бидистиллированной воде и разбавлением;
- раствор нитрата свинца (II) 1х10-2 -1x10-9 моль/л, приготовленный растворением точной навески (чда) в метаноле и разбавлением;
- раствор нитрата свинца (II) 0, 1 - 1 х 10-9 моль/л, приготовленный растворением точной навески (чда) в пиридине и разбавлением;
- ацетатная буферная смесь с pH = 5, приготовленная согласно методике [Флашка];
- раствор нитрата калия 1 моль/л, приготовленный растворением точной навески (хч) в бидистиллированной воде
- раствор уксусной кислоты 0, 1 моль/л приготовленный растворением точной аликвоты (хч) в бидистиллированной воде;
- раствор бензойной кислоты 0, 01 моль/л приготовленный растворением точной навески (чда) в бидистиллированной воде;
- раствор винной кислоты 0, 1 н, приготовленный растворением точной навески (осч) в бидистиллированной воде;
- раствор лимонной кислоты 0, 1 н, приготовленный растворением точной навески (чда) в бидистиллированной воде;
- раствор гидроксида калия 1 - 0, 01 моль/л приготовленный растворением точной навески (чда) в бидистиллированной воде, и разбавлением;
- растворы уксусной (хч), бензойной (чда), винной (осч) и лимонной кислот концентрацией 0, 1 моль/л приготовленные растворением точной навески (аликвоты) в изопропиловом спирте;
- растворы гидроксида калия 0, 2 - 0, 01 моль/л приготовленные растворением точной навески в изопропиловом спирте и разбавлением;
- раствор щавелевой кислоты 0, 1 н, приготовленный из стандарт-титра;
- раствор перманганата калия 0, 1 н, приготовленный из стандарт-титра;
- раствор соли Мора (чда) 0, 1 н, приготовленный растворением точной навески в бидистиллированной воде;
- раствор серной кислоты 1: 4, приготовленный разбавлением;
- раствор трилона Б 0, 01 н, приготовленный растворением точной навески в бидистиллированной воде
- аммиачная буферная смесь с pH = 10, приготовленная согласно методике [Флашка];
- раствор сегнетовой соли 1 моль/л, приготовленный растворением точной навески в бидистиллированной воде;
- трата

1 - бюретка; 2 - ячейка; 3 - магнитная мешалка; 4 - ротор магнитной мешалки; 5 - многопозиционный переключатель; 6 - цифровой вольтметр DT-830B; 7 - ионометр лабораторный pH-150MM; 8 - термодатчик ТДЛ-1000-06; 9 - комбинированный стеклянный электрод ЭСК-10603; 10 - ХСЭ;
11 - сульфидный электрод (PbS); 12 - селенидный электрод (PbSe); 13 - теллуридный электрод (PbTe)
Рисунок 5 - Схема установки для потенциометрического титрования
2. 4 Изучение ионоселективных свойств
2. 5 Изучение индикаторных свойств
2. 5. 1 Кислотно-основное титрование
Поведено потенциометрическое титрование растворов слабых органических кислот различной основности: уксусной, бензойной, винной и лимонной сильным основанием - гидроксидом калия. Именно эти кислоты были выбраны, потому что они часто встречаются в химико-аналитической практике и доступны для большинства современных лабораторий.
В качестве растворителя использовалась дистиллированная вода и изопропанол, а все измерения проводились при температуре 298 К. Объём тит-ранта, отвечающий конечной точке титрования определяли графически по зависимостям ∆E/∆V = ƒ(Vтитранта), для чего строили дифференциальные кривые титрования (рисунки 10-17).

Рисунок 10 - Дифференциальные кривые потенциометрического титрования 0, 1 Н уксусной кислоты 0, 2667 Н гидроксидом калия в водной среде
По полученным данным (рисунок 10) для водной среды можно сделать вывод, что чувствительность к иону H+ увеличивается в ряду PbS <PbSe <PbTe при титровании уксусной кислоты. Скачок потенциала в ктт равен соответственно 0, 148 В для сульфидсвинцового, 0, 244 В для селенидсвинцово-го, 0, 308 В для теллуридсвинцового электродов. Разница между теоретическим объёмом ктт и полученным на халькогенидных электродах не превышает 3, 3 % и минимальна (0 %) для селенидсвинцового электрода.

Рисунок 11 - Дифференциальные кривые потенциометрического титрования 0, 1 Н уксусной кислоты 0, 1482 Н гидроксидом калия в среде изопропанола
Из результатов эксперимента, которые отражены на рисунке 11, можно сделать вывод, что в среде изопропанола, как и в водной среде, чувствительность к H+-иону увеличивается в ряду PbS <PbSe <PbTe при титровании уксусной кислоты сильным основанием (KOH). Скачок потенциала в ктт при титровании в среде изопропанола равен соответственно 0, 008 В для сульфид-свинцового, 0, 184 В для селенидсвинцового, 0, 328 В для теллуридсвинцового электродов. Для сульфид- и селенидсвинцовых электродов скачок потенциала в ктт (∆E) уменьшился в среде изопропанола, по сравнению с водной средой, а для теллуридсвинцового электрода ∆E в изопропаноле незначительно увеличился.
Разница между теоретическим объёмом ктт и полученным на халькоге-нидных электродах отсутствует (0 %), что в свою очередь свидетельствует о большей точности кислотно-основного титрования уксусной кислоты, проводимого в среде изопропилового спирта по сравнению с аналогичным титрованием в водной среде.

Рисунок 12 - Дифференциальные кривые потенциометрического титрования 0, 01 Н бензойной кислоты 0, 03922 Н гидроксидом калия в водной среде
При проведении титрования бензойной кислоты гидроксидом калия в водной среде (рисунок 12) ряд чувствительности к Н+-иону оказался таким же, как и при титровании уксусной кислоты (PbS <PbSe <PbTe). Скачок потенциала в ктт оказался равным соответственно 0, 036 В для сульфидсвинцо-вого, 0, 050 В для селенидсвинцового, 0, 096 В для теллуридсвинцового электродов. Уменьшение ∆E в ктт по сравнению с результатами титрования уксусной кислоты в данном случае связано с тем, что титрованию подвергался более разбавленный раствор бензойной кислоты, что в свою очередь обусловлено низкой растворимостью бензойной кислоты в воде (0, 34 г / 100 мл).
Разница между теоретическим объёмом ктт и полученным на халькоге-нидных электродах не превышает 4 % и максимальна (4 %) для селенидсвин-цового электрода. Большое отклонение ктт, на наш взгляд, связано с ошибкой, допущенной при взвешивании очень малой навески кислоты.
Кроме того, судя по дифференциальной кривой титрования, на которой можно обнаружить несколько незначительных пиков, зафиксированных сульфидсвинцовым электродом, можно сказать, что последний малопригоден для индикации ктт при кислотно-основном титровании не только из-за малого ∆E, но и из-за большого количества шумов.

Рисунок 13 - Дифференциальные кривые потенциометрического титрования 0, 01 Н бензойной кислоты 0, 0101 Н гидроксидом калия в среде изопропанола
Результаты кислотно-основного титрования бензойной кислоты гидроксидом калия в среде изопропилового спирта вновь показали, что чувствительность халькогенидов свинца к присутствию протона водорода в системе увеличивается в ряду сульфид <селенид <;теллурид.
Скачок потенциала системы (∆E) в точке эквивалентности равен: 0, 022 В для сульфида свинца, 0, 132 В для селенида свинца и 0, 180 В для тел-лурида свинца. Вновь наблюдается уменьшение скачка потенциала в ктт, по сравнению с водной средой для PbS и увеличение для PbSe и PbTe.
Разница между теоретическим объёмом ктт и полученным на халькоге-нидных электродах в среде изопропилового спирта отсутствует (0 %), как и при аналогичном титровании уксусной кислоты, а значит среда изопропанола больше подходит для кислотно-основного титрования слабых одноосновных органических кислот с потенциометрическим методом индикации ктт, так как ошибка определения в ней меньше.

Рисунок 14 - Дифференциальные кривые потенциометрического титрования 0, 1 Н винной кислоты 0, 1602 Н гидроксидом калия в водной среде
По полученным данным (рисунок 14) при титровании винной (двухосновной) кислоты в водной среде можно сделать вывод, что чувствительность к иону H+ увеличивается в ряду PbS <PbTe <PbSe, что уже разительно отличается от данных, полученных при титровании одноосновных кислот. Сульфид свинца по прежнему показывает худшие результаты, а вот селенид с теллуридом поменялись местами.
Скачок потенциала в ктт равен соответственно 0, 056 В для сульфид-свинцового, 0, 160 В для селенидсвинцового, 0, 140 В для теллуридсвинцового электродов. Разница между теоретическим объёмом ктт и полученным на халькогенидных электродах не превышает 2 % и минимальна (0 %) для тел-луридсвинцового электрода.
Константы диссоциации винной кислоты (таблица 7) отличаются друг от друга на 1 порядок и согласно [5] различить их методом потенциометрического титрования невозможно. Поэтому небольшой пик на кривой (рисунок 14) в области 11, 5 мл не соответствует теоретической ктт первой ступени (6, 25 мл).

Рисунок 15 - Дифференциальные кривые потенциометрического титрования 0, 1 Н винной кислоты 0, 2963 Н гидроксидом калия в среде изопропанола
Из результатов эксперимента, которые отражены на рисунке 1 5, можно сделать вывод, что в среде изопропанола, в отличии от водной среды, чувствительность к Н+-иону увеличивается в ряду PbS <PbSe <PbTe при титровании винной кислоты гидроксидом калия. Скачок потенциала в ктт по второй ступени при титровании в среде изопропанола равен соответственно 0, 120 В для сульфидсвинцового, 0, 140 В для селенидсвинцового, 0, 200 В для теллуридсвинцового электродов. Для сульфид- и теллуридсвинцовых электродов скачок потенциала в ктт (∆E) увеличился в среде изопропанола, по сравнению с водной средой, а для селенидсвинцового электрода ∆E в изопропаноле незначительно уменьшился.
Разница между теоретическим объёмом ктт полной нейтрилизации и полученным на халькогенидных электродах не превышает 4 %, а максимальное отклонение наблюдается для сульфидсвинцового электрода.
Различить ступени диссоциации винной килоты в среде изопропанола, как и в водной среде, не удалось. Однако, наблюдаемые на рисунке 15 пики в области 5, 5 - 6 мл ближе к теоретической ктт по первой ступени (3, 4 мл).

Рисунок 16 - Дифференциальные кривые потенциометрического титрования 0, 1 Н лимонной кислоты 0, 1602 Н гидроксидом калия в водной среде
Титрование лимонной (трёхосновной) кислоты гидроксидом калия в водной среде показало (рисунок 16), что в используемых условиях (н. у. ) чувствительность к H+-иону увеличивается в ряду PbSe <PbTe <PbS, что значительно отличается от результатов, полученных при титровании одно - и двухосновных кислот в водной среде.
Скачок потенциала в ктт полной нейтрализации (по всем трём ступеням диссоциации кислоты) равен соответственно 0, 270 В для сульфидсвин-цового, 0, 088 В для селенидсвинцового и 0, 176 В для теллуридсвинцового электродов.
Разница между теоретическим объёмом ктт полной нейтрилизации и полученным на халькогенидных электродах не превышает 2 % и минимальна (0 %) для сульфидсвинцового и селенидсвинцового электродов.
Судя по константам диссоциации лимонной кислоты (таблица 7) K1и К3 отличаются друг от друга на 3 порядока, а K1и К2 на 1 порядок и различить их методом потенциометрического титрования очень сложно. Поэтому небольшой пик на кривой (рисунок 16) показанный электродом из сульфида свинца в области 9 мл соответствует теоретической ктт второй ступени (8 мл) с большой погрешностью (11 %).

Рисунок 17 - Дифференциальные кривые потенциометрического титрования 0, 1 Н лимонной кислоты 0, 1482 Н гидроксидом калия в среде изопропанола
Из результатов эксперимента, которые отражены на рисунке 17, можно сделать вывод, что в среде изопропанола, как и в водной среде, чувствительность к Н+-иону увеличивается у PbTe больше, чум у PbSe при титровании лимонной кислоты сильным основанием (KOH). Результаты, полученные на PbS-электроде на графике не приведены, так как их не удалось расшифровать. Это значит, что в рассматриваемых условиях сульфидсвинцовый электрод не может использоваться как индикаторный из-за наличия сильных шумов и большой погрешности определений.
Скачок потенциала в ктт полной нейтрализации лимонной кислоты при титровании в среде изопропанола равен соответственно 0, 076 В для селенид-свинцового, 0, 082 В для теллуридсвинцового электродов. Для теллурид- и се-ленидсвинцовых электродов скачок потенциала в ктт (AE) уменьшился в среде изопропанола, по сравнению с водной средой, что свидетельствует о предпочтительном определении лимонной кислоты в водной среде.
Разница между теоретическим объёмом ктт полной нейтрилизации и полученным на халькогенидных электродах не превышает 3, 3 % и минимальна (0 %) для селенидсвинцового электрода.
По полученным данным для водной среды можно сделать вывод, что чувствительность к иону H+ увеличивается в ряду PbS <PbSe <PbTe при титровании одноосновной кислоты, что согласуется с работой [19]. При титровании двухосновной кислоты ряд изменяется на PbS <PbTe <PbSe, а при титровании трёхосновной на PbSe <PbTe <PbS. В среде изопропилового спирта ряд PbS <PbSe <PbTe сохраняется при титровании кислоты любой основности.
Величина скачка потенциала в точке эквивалентности зависит от материала электрода. В водной среде она уменьшается для PbSe-электрода при увеличении основности кислоты. Для PbS-электрода наибольший скачок потенциала наблюдается при титровании трёхосновной кислоты, а для PbTe-электрода - одноосновной.
В среде изопропанола величина скачка потенциала в точке эквивалентности уменьшается при увеличении основности кислоты для PbSe- и PbTe-электродов. Электрод на основе сульфида свинца в изопропаноле проявляет крайне низкую pH чувствительность, отчего результаты, полученные при потенциометрическом титровании с его использованием часто невозможно интерпретировать.
Таблица 7 - Константы кислотности [Лурье]
|
Кислота |
Ка |
||
|
К1 |
К2 |
К3 |
|
|
бензойная |
6, 17х10-5 |
- |
- |
|
уксусная |
1, 74х10-5 |
- |
- |
|
винная |
9, 1 х10-4 |
4, 3 х10-5 |
- |
|
лимонная |
7, 4х10-4 |
1, 8х10-5 |
4, 0 х10-7 |
По полученным дифференциальным кривым можно различить некоторые константы диссоциации кислот лишь качественно, причём каждая из них в среде изопропанола различима гораздо чётче, чем в водной среде. Например, даже достаточно близкие по значению К1 и К2 винной кислоты отчётливо различимы (рисунки 14 и 15), хотя пики не соответствуют ступенчатой нейтрализации.
Разница между теоретическим объёмом конечной точки титрования и полученным при использовании электродов на основе халькогенидов свинца колеблется от 0 до 4 %.
Таким образом, мембранные электроды на основе теллурида и селени-да свинца могут быть использованы для потенциометрической индикации ктт при кислотно-основном титровании в водной среде и среде изопропилового спирта, а использование сульфидсвинцового электрода в тех же условиях нежелательно.
2. 5. 5 Определение рН-гидроксоосаждения металлов
Определение катионного состава сложных смесей является важной химической задачей. Возможность установление наличия в смеси тяжёлых и токсичных металлов уже давно является важной задачей химии и экологии, как и поиск простых и дешёвых средств для осуществления этой цели.
Практически все гидроксиды тяжёлых и токсичных металлов и некоторых p-элементов нерастворимы в воде и многих других средах и каждый гидроксид осаждается только при определённом уровне pH среды. Поэтому, легко установить качественный состав сложной смеси катионов проведя потенциометрическое титрование сложной смеси щёлочью при постоянной фиксации pH среды.
Для апробации возможности использования изготовленных электродов на основе халькогенидов свинца в подобных измерениях проведено потенциометрическое титрование 50 мл водного модельного раствора, содержащего по 0, 01 моль/л следующих катионов: Ca2+, Pb2+, Fe3+, Zn2+, Cu2+, Al3+, Co2+, подкисленных азотной кислотой для предотвращения гидролиза. По результатам титрования были построены графические зависимости ∆E/∆V = f(pH), которые можно найти в приложении Б. На рисунках 30-32 представлены гистограммы, отражающие скачок потенциала на халькогенидном электроде при достижении pH осаждения каждого из катионов металлов, содержащихся в смеси.

Рисунок 18 - Скачок потенциала на сульфидсвинцовом электроде при pH осаждения катионов металлов

Рисунок 19 - Скачок потенциала на селенидсвинцовом электроде при pH осаждения катионов металлов

Рисунок 20 - Скачок потенциала на теллуридсвинцовом электроде при pH осаждения катионов металлов
При анализе исследуемого модельного раствора каждый из электродов оказался нечувствительным к осаждению одного из семи катионов, содержащихся в смеси. На сульфиде и теллуриде свинца не ужалось зафиксировать осаждение Co(OH)2, а на селениде свинца Al(OH)3. Кроме того, на всех электродах удалось зафиксировать растворение гидроксидов цинка и алюминия. В таблице 10 представлены сравнительные данные по pH-осаждения гидроксидов металлов, полученные на испытуемых электродах.
Для изучения отклонения полученных данных от теоретических значений были построены зависимости рНтеория = ƒ(рНпрактика) представленные на рисунке 21, на котором курсивом отмечена теоретическая прямая.
Таблица 10 - Сравнение значений pH-гидроксоосаждения, полученных на халькогенидных электродах с теоретическими
|
Гидроксид |
pHосаждения |
|||
|
теория |
PbS |
PbSe |
PbTe |
|
|
Fe(OH)3 |
2, 30 |
2, 65 |
2, 86 |
2, 11 |
|
Al(OH)3 |
4, 00 |
4, 22 |
- |
3, 83 |
|
Cu(OH)2 |
5, 30 |
5, 40 |
4, 80 |
5, 28 |
|
Pb(OH)2 |
6, 00 |
5, 90 |
6, 14 |
6, 14 |
|
Zn(OH)2 |
6, 40 |
6, 30 |
6, 35 |
6, 35 |
|
Co(OH)2 |
7, 60 |
- |
8, 01 |
- |
|
Ca(OH)2 |
12, 40 |
12, 32 |
12, 21 |
12, 3 |

Рисунок 21 - Зависимости pHтория = ƒ(pHпpaкткa) ДОя сульфидсвинцового (а), селенидсвинцового (б) и теллуридсвинцового (в) электродов
Список использованных источников
1. Лопатин, Б. А. Теоретические основы электрохимических методов анализа: учеб. пособие для ун-тов / Б. А. Лопатин. - М.: «Высшая школа», 1975. - 295 с.
2. Плэмбек, Дж. Электрохимические методы анализа: пер. с англ. / Дж. Плэмбек. - М.: Мир, 1985. - 496 с.
3. Варламова, И. А. Физико-химические методы анализа: учеб. пособие / И. А. Варламова, Л. Л. Калугина, Л. Г. Коляда. Магнитогорск: Магнит. гос. техн. ун-т, 2008. - 67 с.
4. Денисова, Г. П. Электрохимические методы анализа. Электропроводящие полимерные композиции: учеб пособие / Г. П. Денисова, А. В. Денисов, С. С. Попова. Саратов: Сарат. гос. техн. ун-т, 2009. - 52 с.
5. Будников, Г. К. Основы современного электрохимического анализа / Г. К. Будников, В. Н. Майстренко, М. Р. Вяселев. - М.: Мир: Бином ЛЗ, 2003. - 592 с.
6. Кокшарова, И. У. Электрохимические методы анализа: учеб. пособие / И. У. Кокшарова. Волгоград: ВолгГТУ, 2003. - 55 с.
7. Шрайбман, Г. Н. Потенциометрическое титрование: методические указания / Г. Н. Шрайбман, Н. В. Серебренникова, П. Д. Халфина. - Кемерово: Кемеровский государственный университет, 2004. -40 с.
8. Ковганко, В. Н. Физико-химические методы анализа: тексты лекций / В. Н. Ковганко. Минск: БГТУ, 2009. - 104 с.
9. Зятьков, И. И. Сенсоры на основе полевых транзисторов: учебное пособие / И. И. Зятьков, А. И. Максимов, В. А. Мошников. - СПб.: Изд-во СПбГЭТУ «ЛЭТИ», 2002. - 56 с.
10. Мидгли, Д. Потенциометрический анализ воды / Д. Мидгли, К. Торренс; пер. с англ. Б. Г. Кахана. - М.: Мир, 1980. - 519 с.
11. Каттралл, Р. В. Химические сенсоры / Р. В. Каттралл; пер. с англ. О. О. Максименко. М.: Научный мир, 2000. - 144 с.
12. Камман, К. Работа с ионселективными электродами / К. Камман; пер. с нем. А. Ф. Жукова. - М.: Мир, 1980. - 285 с.
13. Эггинс, Б. Химические и биологические сенсоры / Б. Эггинс; пер. с англ. М. А. Слинкина. М.: Техносфера, 2005. - 336 с.
14. Корыта, И. Ионоселективные электроды / И. Корыта, К. Штулик; пер. с чешск. А. Р. Тимербаева. - М.: Мир, 1989. - 272 с.
15. Байулеску, Г. Применение ион-селективных мембранных электродов в органическом анализе / Г. Байулеску, В. Кошофрец; пер. с англ. В. В. Соболя. - М.: Мир, 1980. - 231 с.
16. Никольский, Б. П. Ионоселективные электроды / Б. П. Никольский, Е. А. Матерова. - Л.: Химия, 1980. - 240 с.
17. Hirata, H. Lead sulfide-impregnated silicone rubber membranes as selective electrodes for lead ion / H. Hirata, К. Date // Analytical chemistry, Vol. 43, № 2, 1971. Pp 279-281.
18. Hirata, H. Analytical study of the lead ion-selective ceramic membrane electrode / H. Hirata, К. Higashiyama // Bulletin of the Chemical Society of Japan, Vol. 44, № 9, 1971. Pp 2420-2423.
19. Hansen, E. M. Selectrode - the universal ion-selective electrode: Part VIII. The solid-state lead (II) selectrode in lead (II) buffers and potentiometric titrations / E. M. Hansen, J. Ruzicka //Analytica Chimica Acta, Vol. 72, № 2, 1974. Pp 365-373.
20. Лакшминараянайах, Н. Мембранные электроды / Н. Лакшминараянайах; пер. с англ. В. А. Станкевича и И. С. Ивановской. Л.: Химия, 1979. - 360 с.
21. Hirata, H. Ion-selective lead selenide and lead telluride membrane electrodes / H. Hirata, K. Higashiyama // Analytica Chimica Acta, Vol. 57, № 2, 1971. Pp 476-477.
22. Majer, V. Behaviour of Lead Ion Sensitive Chalcogenide Electrodes in Buffered Lead-Ion Solutions / V. Majer, J. Vesely, K. StuHk // Analytical Letters, Vol. 6, Issue 6, 1973. Pp 577-584.
23. Vesely, J. Titration of sulphates and lead using lead ion selective electrode / J. Vesely // Collection of Czechoslovak Chemical Communications, Vol. 46, № 2, 1981. Pp 368-376.
24. Rechnitz, G. A. Potentiometric measurements in aqueous, non -aqueous, and biological media using a lead ion-selective membrane electrode / G. A. Rechnitz, N. C. ^nny // Analytical Letters, Vol. 3, Issue 5, 1970. Pp 259-271.
25. Воган, Д. Химия сульфидных минералов / Д. Воган, Дж. Крейг; пер. с англ. Н. С. Бортникова, Р. М. Минеевой. - М.: Мир, 1981. - 575 с.
26. Хариф, Я. Л. Свинца халькогениды / Я. Л. Хариф, П. В. Ковтуненко // ChemPort. Ru. - Режим доступа: http: //www. chemport. ru/data/chemipedia/article_3361. html
27. Халькогениды и оксиды элементов IV группы. Получение, исследование, применение / О. А. Александрова, А. И. Максимов, В. А. Мошников, Д. Б. Чеснокова; ООО «Технолит». - СПб.: Изд-во «Технолит», 2008. - 240 с.
28. Самсонов, Г. В. Сульфиды / Г. В. Самсонов, С. В. Дроздова. -М.: «Металлургия», 1972. - 304 с.
29. Чижиков, Д. М. Селен и селениды / Д. М. Чижиков, В. П. Счастливый. - М.: «Наука», 1964. - 322 с. 5
30. Чижиков, Д. М. Теллур и теллуриды / Д. М. Чижиков, В. П. Счастливый. - М.: «Наука», 1966. - 278 с. 6
Скачать диплом: